基因疗法改写“被诅咒”的命运,杜氏肌营养不良治疗现曙光
 医学资讯
|
阅读量:次
医学资讯
|
阅读量:次
据统计,全球平均每3500个男婴中就有一个患有杜氏肌营养不良这种可怕的病。患者一般在3-5岁开始发病,早期表现出进行性腿部肌无力(爬楼梯困难),此后进行性加重,12岁时失去行走能力, 需要借助轮椅。随着疾病的进展,患者可能会出现危及生命的心肌病或呼吸衰竭,导致早逝(通常是二三十岁)。
杜氏肌营养不良(简称DMD)是一种致命的神经肌肉疾病,是由X染色体上dystrophin基因突变引起的。这种突变导致患者肌肉中缺少抗肌营养不良蛋白(dystrophin),而这种蛋白能保持肌肉细胞完整,一旦缺乏就会导致进行性肌肉退化和萎缩。
像大多数罕见的遗传性疾病一样,DMD目前无法治愈。美国FDA曾在2016年批准了首款治疗DMD的药物Exondys 51 (Eteplirsen) ,用于大约13%已明确51号外显子突变的DMD患者。2019年12月FDA加速批准了Vyondys 53 (Golodirsen)注射剂用于确诊为发生53外显子跳跃基因突变的DMD患者。
针对DMD等罕见病,科学家致力于从基因水平上改变其治疗,目前最领先的是基因疗法SRP-9001(也称:AAVrh74.MHCK7.micro-Dystrophin) 。该疗法将MHCK7启动子与AAVrh74载体结合,以提供设计用于维持血影蛋白样重复序列2和3的微型抗肌营养不良蛋白转基因。目前,SRP-9001正在进行临床研究Study-101。
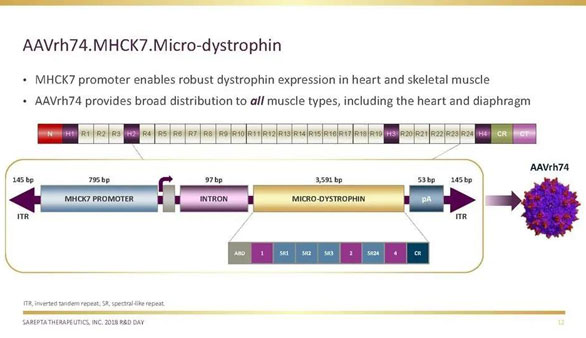
▲图片来源:Sarepta
微型抗肌营养不良蛋白(micro-dystrophin)基因治疗显疗效
日前,美国国家儿童医院的Creative Commons研究人员已在JAMA Neurology上发表了针对DMD患儿进行系统性微营养不良蛋白(micro-dystrophin)基因疗法的首期临床试验结果。最新结果显示,相比标准治疗,SRP-9001基因疗法带来的功能改善更为显著。
常规的基因治疗方法是利用病毒载体向体内递送功能性基因拷贝,但对于DMD这一疾病而言,缺失的抗肌营养不良蛋白基因太长,难以使用常规的载体工具进行递送。为此研究人员开发了micro-dystrophin作为一种微基因,它在提供功能的同时仍然适合载体。
Study-101(NCT03375164)试验招募了4名患有DMD的男孩,年龄在4到7岁之间。他们通过外周静脉注射了2.0 x 1014 vg / kg 的单剂量SRP-9001。所有治疗相关事件均为轻至中度,无严重不良事件发生。最常见的治疗相关不良事件是呕吐(18例中的9例)。三名患者的γ-谷氨酰转移酶水平暂时升高,可通过糖皮质激素缓解。

输注12周后,腓肠肌活检标本显示,平均81.2%的肌纤维表达微抗肌营养不良蛋白,肌膜处平均表达强度为96%。蛋白质印迹显示平均表达为74.3%(未调整脂肪或纤维化)和95.8%(调整后)。
所有参与者均已确认载体转导,NSAA评分(一个17项评定量表,用于测量运动能力,评分范围为0到34)显示运动功能改善。所有参与者也显示肌酸激酶(CK)水平下降,维持了一年。
其他功能结果测量包括起身时间、爬四层楼梯、100米计时测试以及对膝关节伸屈肌、肘关节伸屈肌和屈肌的手扶动力测量。
Mendell博士说:
“杜氏肌营养不良症很难治疗,基因疗法提供了可能改变疾病进程的必要选择。临床试验结果验证了该疗法的安全性及有效性,也为后续的临床试验提供了概念验证支持。”
该研究计划持续到2021年3月,期待它能早日获批,彻底改变DMD患者的生活!
2020-06-17 15:14

好医友小编
联系医学顾问
医学顾问微信在线

 友情链接
友情链接
 公司地址
公司地址
中国总部:浙江省杭州市下城区永福桥路17号汉鼎国际大厦北楼19层
美国总部:100 W Villa Street, Suite 101,Pasadena, CA 91103
厦门分部:厦门市思明区云顶中路515号C幢8楼
北京分部:北京市朝阳区望京宏泰东街绿地中国锦25层
 免费咨询 400-886-0922
免费咨询 400-886-0922

关注我们,查看更多资讯

 咨询医学顾问:400-886-0922
咨询医学顾问:400-886-0922